


Chemical journal publishes JNU research paper
Time: July. 21st, 2018
Source: College of Science and Engineering
Chen Xiaobo, an associate researcher in JNU's College of Science and Engineering, and Chen Shiqi, a graduate student, have published a research paper in the international journal of the American Chemical Society. The paper, titled Reaction Mechanism with Thermodynamic Structural Screening for Electrochemical Hydrogen Evolutionon Monolayer1T' phase MoS2,” was published on July 8 in Chemistry of Materials (IF = 9.89). Chen Xiaobo is the project leader, and Chen Shiqi is the first author.
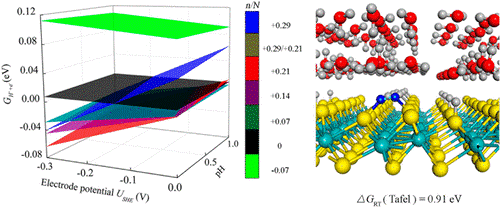
This research gives a totally new idea about the Volmer-Heyrovsky kinetics mechanism of hydrogen precipitation, which has the transition-metal sulfide surface that uses MoS2 as the main research object. It also makes the point that hydrogen evolution reaction relies on the diffusion-assisted Volmer-Tafel mechanism.
Abstract:
We report on a density functional theory (DFT) calculation to determine the reaction pathway and barrier of the hydrogen evolution reaction (HER) at the interface of monolayer 1T′ phase MoS2 and water. By screening the interfacial structures with the lowest chemical potential of protons and electrons, key structural characteristics that are important for prediction of reaction mechanism are identified. Under typical reaction conditions of HER, the catalyst surface features a high proton coverage of ca. 37% while the aqueous solution has a relatively low hydronium concentration of no more than ca. 1.8%. This contrast leads to proton desorption from the catalyst surface through a diffusion-assisted Tafel manner, rather than the Heyrovsky manner assumed previously. The result is supported by the agreement of the calculated reaction barrier and surface coverage with those of experimental estimate. In prediction of catalytic activity, hydrogen adsorption energies of reaction intermediates are widely used as the thermodynamic descriptor, while reaction barriers usually serve as kinetic parameters. We suggest that both thermodynamic and kinetic description toward HER should be performed on the premise that the lowest chemical potential of protons and electrons is obtained.
(Figure 1. (a) Side and (b) top view of an electrochemical double layer (EDL) interfacial model consisting of monolayer 1T′phase MoS2 and two layers of water molecules. Two categories of sulfur atoms labeled by S-I and S-II are identified on the catalyst surface, which differs from each other in the normal height. The rectangular 2 × 2 super cell lined out by the black box in (b) is used for DFT calculations, which consists of 8 MoS2 formula units and 14 water molecules.)
Link to paper:https://pubs.acs.org/doi/10.1021/acs.chemmater.8b02236
NEWS
- About the University
- Quick Links
Copyright © 2016 Jinan University. All Rights Reserved.